





- AllProduct Category
-
Purification Engineering
Air Shower Room
Cleanroom Workbench
Transfer Window
Air curtain












详情描述
Medical Device GMP Manufacturing Facility
Applicable Industries: Sterile Medical Devices, Medical Supplies
Suggested Grade: 100th Level - 300,000th Level




























.jpg)



Medical Equipment GMP Manufacturing Facility Solution:
1. Implanted medical devices where the contamination of active substances, inactivated substances (including pyrogens) significantly affects the product require GMP车间 planning and design for medical device manufacturing. Control of the working environment should be implemented, and the sterilization methods should be validated and documented. The production and packaging of such products should be carried out in a regulated, controlled environment (GMP facility).
2. For non-sterile implantable medical devices or medical devices intended to be sterilized before use, if the product's cleanliness and packaging processes can reduce contamination and maintain consistent control levels through verification, a controlled environment should be established in the GMP workshop for medical devices to contain the verified cleanliness and packaging processes. Manufacturers may refer to the YY0033-2000 standard or independently verify and determine the product's production cleanliness level.
3. Implement control measures for products that are contaminated or prone to contamination. Establish documentation for handling, cleaning, and decontamination procedures for contaminated or easily contaminated products, work surfaces, or personnel.
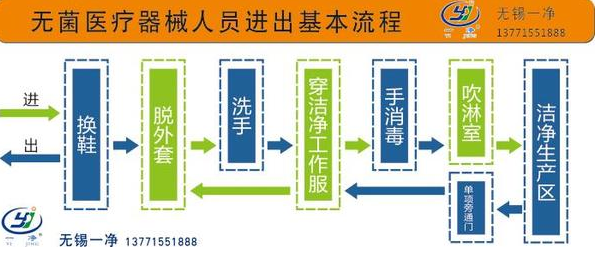
2010 Revised GMP Cleanroom Classification
Grade A: High-risk operation areas, such as filling zones, areas for placing stopper drums, and open-mouth packaging containers in direct contact with sterile preparations, as well as areas for sterile assembly or connection operations, should maintain the environmental status with unidirectional airflow benches (shelters). The unidirectional airflow system must provide uniform air supply within its working area at a speed of 0.36-0.54 m/s (guideline value). Data proving the status of unidirectional airflow should be available and verified. Lower air speeds may be used within enclosed isolation devices or glove boxes.
Grade B: Refers to the background area surrounding Grade A cleanroom where high-risk operations such as sterile preparation and filling are performed.
C and D Grades: Refers to the clean areas of operation with lower importance in the sterile pharmaceutical production process.

China GMP (2010 Revision) 2010 Edition, Annex 1 of GMP, Chapter 3, Article 9...The standard specifications for air-borne particles in different grades of clean areas are as follows in the table:
(1) To confirm the grade of Grade A clean area, the sampling volume at each sampling point must not be less than 1 cubic meter. The level of airborne particles in Grade A clean area is ISO 4.8, with a limit standard of ≥5.0μm suspended particles. The level of airborne particles in Grade B clean area (static) is ISO 5, including both particle sizes listed in the table. For Grade C clean area (both static and dynamic), the levels of airborne particles are ISO 7 and ISO 8, respectively. For Grade D clean area (static), the level of airborne particles is ISO 8. The testing method can refer to ISO 14644-1.
(2) When confirming the grade, a portable dust particle counter with a shorter sampling tube should be used to prevent particles ≥5.0μm from settling in the long sampling tube of the remote sampling system. In unidirectional airflow systems, a sampling head with isokinetic properties should be employed.
(3) Dynamic testing can be conducted during routine operations and simulated culture medium filling processes to demonstrate achievement of dynamic cleanliness levels; however, dynamic testing during simulated culture medium filling trials must be conducted under "poor conditions."

Regulatory references for GMP车间 engineering design of medical equipment:
1. International Standard ISO/DIS 14644
2. Cleanroom Factory Design Standard GB50073-2001
3. Medical Device Packaging Workshop Clean Room Factory Standard (GMP-97)
4. Good Manufacturing Practices (GMP) - 98 for Pharmaceutical Production
5. Cleanroom Construction and Non-Collectible Standard (JGJ 71-90)
6. Ventilation and Air Conditioning Construction and Acceptance Standard GB 50243-2002
7. Federal Standard FS209E-92 of the United States

Medical Equipment GMP Workshop Construction Design Reference:
1. "Medical Device Production Enterprise Quality Management Specification (Trial) by the National Food and Drug Administration (2009) -- Abolished in 2015."
"Regulations for the Production of In-Vitro Diagnostic Reagents (Trial Version), National Food and Drug Administration (2007) -- Abolished in 2015."
Notice on the Implementation of the "Temporary Regulations for the Quality Management System of Medical Device Production" and Its Supporting Documents (2011) -- Abolished in 2015.

Additional Clarification: Net Purification
The regulations, specifications, and standards for medical devices from 2007 and 2009 were discontinued in 2015. In their place, the 2015 Quality Management Specifications for Medical Device Manufacturers, along with three appendices on sterile, implantable, and in vitro diagnostic reagents, replaced the planning for GMP workshops for medical devices.
5. "Standard for the Production Management of Sterile Medical Devices" (YY0033-2000)
6. "Clean Factory Design Code (GB50073-2010)"
7. "Cleanroom Construction and Acceptance Standards" (GB50591-2010)
……
19. "Part 1: General Requirements for Sterilization of Medical Products" (YY/T0567.1-2005)
20. "Outline of Sterile Medical Device Production and Quality Management," National Medical Products Administration (2000)
"23. 'Quality Control and Evaluation of Sterile Medical Devices,' published by Soochow University Press (2012)"
24. "Aseptic Medical Device Production and Cleanroom Construction", CMD (2009)

Consider the following issues in the construction of medical equipment purification projects:
Cleanroom materials required for the packaging workshop of medical equipment.
2. Comprehensive services for the design, installation, commissioning, and maintenance of cleanrooms in medical equipment manufacturing facilities and packaging workshops.
3. Cleanroom HVAC Purification System for Medical Device Packaging Workshop
a. Cleanroom Temperature and Relative Humidity:
Sterile medical devices typically require temperatures within the range of 18~28°C as per regulatory standards, with humidity levels between 45%~65%. Most companies can maintain these conditions. If, during dynamic monitoring, the requirements are not met, it may indicate the presence of heat-generating equipment indoors.
B. Cleanroom air volume, air change rate, static pressure difference:
In a cleanroom, the number of air changes is determined by the room's supply air volume, while the static pressure difference depends on the difference between the supply, return, and exhaust air volumes. The total supply air volume, fresh air volume, total exhaust air volume, and external pressure difference can be achieved by adjusting the fan speed or the total valve opening. The air volume and pressure in each room can be controlled by adjusting the branch pipeline valve opening.
C. Cleanroom suspended particles, airborne bacteria, settled bacteria:
If the test conditions cannot meet the specified environmental parameters (temperature and humidity, wind speed, air changes, and static pressure difference within the specified range), the test results of critical items such as suspended particles, airborne bacteria, or settling bacteria should be considered invalid. Since temperature, relative humidity, wind speed, air changes, and static pressure difference together constitute the microclimate of a clean room and are important indicators of the normal maintenance of the clean room, the test of critical items in key processes can be revised to a comprehensive performance test of all processes in key processes. Only in this way can a comprehensive and systematic monitoring of the production clean room be achieved. To ensure the scientificity and accuracy of the data for monitoring the performance of the clean room, the testing department should simultaneously conduct tests of temperature, relative humidity, air changes, and static pressure difference as preconditions when performing critical item suspended particle and microbial tests.
d. Cleanroom Temperature:
The reason for the room temperature in cleanrooms exceeding the designed range during summer is often due to the initial determination of air conditioning supply volume and air change rate for each cleanroom, which only focuses on meeting cleanliness standards while neglecting the verification and calculation of thermal balance in each cleanroom. Therefore, during the design and operation of cleanrooms for production, it is essential to make real-time corrections to the air conditioning supply parameters to ensure that the temperature in cleanrooms is maintained between 18-28℃ throughout all seasons. Temperature and relative humidity mainly affect product production processes and bacterial propagation conditions, and can also trigger the impact of operator comfort on product quality.
e. Cleanroom air supply volume, air changes per hour:
Medical Equipment Purification Engineering - In the design phase of aseptic cleanroom engineering, the determination of the air supply volume must first meet the air change requirements of the corresponding cleanliness level, and then further determine the air volume through thermal and humidity load checks. On this basis, the selection of high-efficiency filters is carried out. The air volume handled by the filters should be less than or equal to the rated air volume, and the resistance and efficiency of high-efficiency (sub-high efficiency, ultra-high efficiency) air filters set in the same cleanliness area should be similar.

General Requirements for GMP Compliance in Medical Device Manufacturing Facilities:
(1) Smooth surface; (2) Durable surface; (3) Good thermal insulation; (4) Low static electricity generation; (5) Non-absorbent and moisture-proof; (6) Excellent sound absorption.
(7) Easy to process; (8) Resists dust accumulation on the surface; (9) Dust can be easily removed.






.jpg)
询价单


